|
Disclaimer:
The primer design program accessed by this site is provided "as is" by the British Columbia Cancer Agency's Genome Sciences Centre. Any express or implied warranties of fitness for a particular purpose are disclaimed. Acknowledgments:
Software: Primer design is automated
using
a PERL program
that does the following:
AcePerl
is used to extract gene information and DNA sequence from the
C. elegans
AceDB database. This
information
is used to build a map of gene and DNA sequence features that is used to
design primers that flank the gene or specific exons within the gene while
avoiding problem areas such as overlapping genes or repeat
sequences.
Optimal primer sequences are predicted using Primer3
(developed by the Whitehead Institute for Biomedical Research).
Primer
sets are evaluated using the primer pair penalty (Q-value) assigned by
Primer3 and by testing for multiple or unwanted PCR products with
"electronic
PCR" (e-PCR).
Gene Name: The 3-letter locus name or predicted gene name (for example C02D5.1). You can find information on known or predicted C. elegans genes in wormbase. Primer sets per exon: Defines how many sets are designed for each exon in the gene. The programs walks through the gene exon by exon until primers have been designed to flank each one. The default value is five sets for each exon. In cases where the same set of primers is predicted for multiple exons, redundant sets are skipped to reduce computational load. Coordinates: Specify the coordinates of the PCR target. For gene-based primer design of a particular exon, use Target exon. Target exon: Restrict the primer design to a particular exon. If you are interested in a specific region of the gene or are trying to single one of a number of multiple splice products, enter the exon number in the "target exon" box. Leaving it blank will result in a set of primers for each exon. PCR product: The desired size of the external (first-round) PCR product. The larger the size range, the higher the chances of finding the best possible PCR primers. We use about 3 kb for the standard knockout protocol and about 2 kb for the poison primer approach. Primer size: The desired oligonucleotide length. The default is 20. In most cases the size range is not critical because Primer3 is biased towards selecting primers at the user-defined optimum size. Optimum Tm: The desired oligonucleotide melting temperature. The default is 60 degrees centigrade. In most cases the temperature range is not critical because Primer3 is biased towards selecting primers at the user-defined optimum value. External/internal primer interval: This means the maximum allowed distance between the external (first-round) primers and the internal (nested) primers. The default is 200 bp. The larger this size range, the better the chances of getting good primers. This primer design program is meant for gene-knockout primer sets but is also useful for more general purpose PCR. If you do not want nested primers, simply leave this box empty. GC clamp: The number of G or C bases that must be at the 3' end of the primer. Maximum Q value: This subject is discussed below. Allow primers in neighboring genes: Checking this box will allow primers to be designed in sequences contained in adjacent or overlapping genes. It may be necessary to do this if you need primers for the first or last few exons when the neighbors are too close. Poison primers: Selecting poison primers will design one (left or right) or two (both) primers that are designed to pair with one or both of the external PCR primers. When the poison primer(s) are included in the external PCR reaction, they cause a truncated PCR product that can out-compete the full-length product and interfere with the second-round amplification. The purpose of the poison primer approach is to allow the detection of small deletions or deletions that would otherwise have been missed in a complex mixture. Complete the "separation" box to define the maximum allowed distance between the 5' ends of poison primers. Restricting this distance too much will make it difficult to find a complete set of primers due to the limited amount of sequence available in a small interval. cDNA or RT-PCR primers: Check this box to prevent primers from being located in non-transcribed DNA. This feature can be used to design primers for reverse transcriptase PCR or to amplify cDNAs. Note that this feature is not compatible with nested internal primers or poison primers. The suggested size range is 200 to 1000 bp. See also: PCR strategy and a detailed description of the poison primer technique.
PCR Strategy:
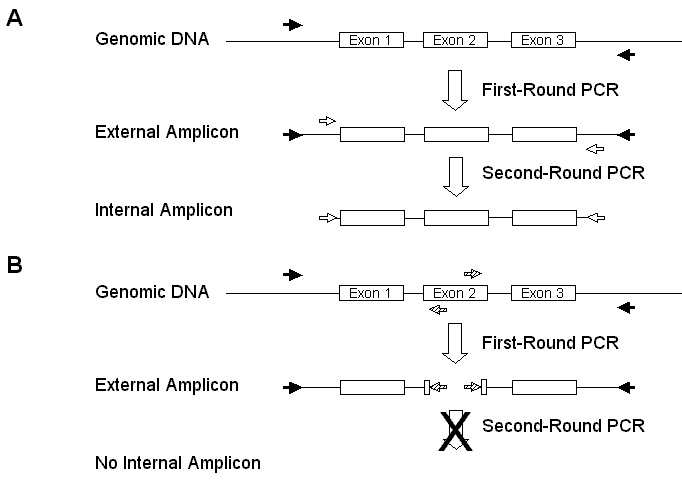
Figure 1. Polymerase Chain Reaction (PCR) based screening
strategies for deletions affecting C. elegans genes. A) Standard
screening strategy. PCR amplification of the gene of interest in
done in two steps. The first-round PCR with external primers
produces
an amplicon that contains all or part of the gene. The external
amplicon
is re-amplified in a second-round PCR using nested internal primers.
The internal amplicon in analysed with agarose gel electrophoresis.
Deletions are detected by a reduction in the size of the internal
amplicon.
B) .Poison Primer. screening strategy. PCR is done as above
except that one or two additional primers are introduced into the first
round amplification. Competitive effects in the multiplex PCR result
in the reduction or elimination of the full-length external
amplicon.
Second round amplification with the internal primers is inhibited by the
poisoning of the first round amplification. Deletions that remove
the target exon (the poison primers binding site) restore amplification
of the external amplicon and allow the second-round amplification to
proceed
normally, resulting in the detection of an internal amplicon.
Primer Quality:
This number is the penalty for how far the primer pairs depart from
optimality
criteria. Lower is better.
A value shown in red indicates that the user-defined maximum penalty has
been exceeded.
In the default primer3 settings, deviations from optimum
primer size and Tm have a large influence on this value. Go here
for more information on how Primer3 assigns the Q value. It is more
difficult to find good quality nested primers
or poison primers because of the limited amount of available
sequence.
It may help to adjust parameters such as PCR product size, primer size
and the allowed distance between external and nested primers.
The gene knockout primer sets are designed to aid in identifying deletions that produce null mutations by removing coding DNA. The coding DNA field in the PCR product information table refers to the number of bases within the PCR product that are exonic sequence, or genomic DNA sequence that forms part of the mRNA that is ultimately translated into amino-acid sequence. Non-coding DNA, such as intronic or flanking sequences, is not included in this calculation. The "portion of gene" field refers to the proportion of the total
coding
DNA for the gene that is contained within the PCR product.
Non-coding
DNA sequence is not included in this calculation. For example, if
a PCR product contained 200 bp of coding DNA, and the mRNA for the entire
gene contains 600 bp of coding DNA, the PCR product is said to contain
33% of the gene.
The coordinates shown for PCR primers refer the the position of the 5' end of the primer, regardless of its orientation. The size of the PCR product is simply the difference between the two 5' coordinates. Typically, the reference sequence is the source clone for the gene. For example the predicted gene C02D5.1 uses the sequence coordinates of the cosmid clone C02D5 as its reference. In cases where the gene or the PCR primers span the end of a clone, a higher order coordinate system is employed. For the C. elegans genome in AceDB, this usually means a Link or Superlink sequence. Here is where we use e-PCR to scan the entire C. elegans genome to look for potential false priming and spurious PCR products. The word size and allowed mismatch parameters affect the stringency of predicted primer binding. Word size means how much of the 3' end of the primer has to be a perfect sequence match. Allowed mismatches indicates how many nucleotide mismatches are allowed in the remaining (5') portion of the primer. We use default values of W=7 (perfect sequence match for the seven nucleotides at the 3' end of the primer) and N=2 (two mismatches allowed in the remaining portion of the primer). This means that any part the DNA sequence in the C. elegans genome that contains a region where there is a perfect match for the 3' end of the primer and two or fewer mismatches in the 5' end will be called as a good primer binding site. For the external PCR all possible primer pairs are evaluated to detect
potential false priming. The entire C. elegans genome and
(for nested primers) the external PCR amplicon are searched for the
following
pairs:
Scanning for false priming can be quite time consuming if a large
number
of primer sets is being considered. Choosing the "test selected
primers"
option will allow you to test only those primers that suit your needs
without
wasting time on sets that do not.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||